Печень
Несколько слов о красящих веществах - пигментах, играющих важную роль в нашем теле. Важнейшие из них гемоглобин и миоглобин. Эти химические вещества - очень близкие родственники: гемоглобин - красящее вещество крови, миоглобин - мышц. Белковая часть гемоглобина носит название глобин, а красящая часть - гем. Гем представляет собой органическое вещество протопорфирин, в состав которого входит один атом железа. Именно гем обладает способностью связывать кислород и легко отдавать его. Благодаря этому гемоглобин выполняет свою основную работу - доставку кислорода от легких к тканям. Гем содержится также в цитохромах и, следовательно, помогает клеткам использовать кислород. Он входит в состав фермента - каталазы, участвующей в процессах окисления. Миоглобин, содержащий гем, тоже дыхательный пигмент, обеспечивающий в мышцах кратковременный запас кислорода.
Распад гемоглобина может происходить в нашем теле всюду, даже если он выходит за пределы кровеносной системы (синяки, которые иногда возникают при ушибах). Но главную работу по разложению гемоглобина ведут клетки печени: они превращают гемоглобин в желчные пигменты, главным образом билирубин.
Печень напоминает огромную центральную лабораторию, в различных уголках которой напряженно трудится целый коллектив химиков, выполняющих самую разнообразную работу. Она активно вмешивается в самые интимные стороны химических превращений обмена веществ в нашем теле и стоит на страже его безопасности. Как же это происходит? Печень, расположенная на пути тока крови от ворсинок кишечника ко всем органам и тканям, в значительной мере перераспределяет содержание в организме углеводов, жиров и белков и их использование.
Начнем с углеводов. Печень чутко реагирует на запросы органов и тканей: при недостатке углеводов она использует свой запас гликогена, превращая его в глюкозу. Справиться с огромной разветвленной молекулой этого полисахарида - дело нелегкое, и печень осуществляет это двумя путями. Основной путь - фосфоролиз (расщепление при помощи фосфора). Ведает этим путем фосфорилаза - фермент, содержащийся в печени в двух формах - активной и пассивной. Действие активной фосфорилазы заключается в разрушепии молекулы гликогена и превращении его в глюкозу, соединенную с фосфорной кислотой - глюкозо-6-фосфат. Этот фермент работает в содружестве с другим ферментом, который как бы готовит рабочее место, очищая путь для воздействия фосфорилазы на гликоген. Но чтобы поддержать уровень сахара в крови, необходима свободная глюкоза, которая и освобождается из глюкозо-6-фосфата под действием специального фермента.
Печень может разрушать гликоген и другим путем - с помощью фермента амилазы, похожего на ту амилазу, которая в основном расщепляет полисахариды в процессе пищеварения. (Она выделяется со слюной и с соком поджелудочной железы.) Но, как мы уже упоминали, этот путь превращения гликогена в глюкозу не главный.
У печени есть и другие ферменты. Ведь в состав нашей пищи входят и такие вещества, которые содержат наряду с глюкозой и простые сахара - фруктозу (в обычном пищевом сахаре) и галактозу (в молочном сахаре) В случае необходимости печень задерживает эти моносахариды и превращает их с помощью специальных ферментов в глюкозу. Печень строит молекулу глюкозы и из таких веществ, которые легко образуются при распаде этого сахара (молочная кислота, глицерин и др.). Но печень не ограничивается построением глюкозы заново. Давно уже замечено, что больные сахарной болезнью могут выделять глюкозу и тогда, когда пища совершенно не содержит углеводов. Ученые сразу же заподозрили в этом печень. Так и оказалось. Печень может превращать в глюкозу самые разнообразные вещества и даже такие, которые содержат азот, в частности разные аминокислоты - глицерин, аланин, серии и др. Теперь остановимся на некоторых нарушениях обмена, которые можно связать с деятельностью печени.
Фруктозурия представляет редкое расстройство обмена, при котором плохо используется и выделяется с мочой фруктоза. Тип наследования аутосомно-рецессивный. Существуют две формы фруктозурии. Одна доброкачественная, которую ошибочно можно принять за диабет. Частота распространения этой формы фруктозурии приблизительно 1 случай на 130 тыс. человек.
Место ошибки обмена при доброкачественной фруктозурии не установлено. Начинается обмен фруктозы с превращения ее в фруктозо-1-фосфат. В печени этим превращением ведает специальный фермент фруктокиназа. Очевидно, именно здесь происходит ошибка обмена, и недостаток фруктокиназы влечет фруктозурию. Возможно, что большую роль играет альдолаза, недостаток которой мешает использованию образовавшегося фрукто-зо-1-фосфата (рис. 38). Точных сведений пока нет. Однако имеются основания полагать, что дефект этого фермента (точнее, фруктозо-1-фосфатальдозы) обусловливает качественно другую, гораздо более тяжелую и более редкую форму фруктозурии, тоже аутосомно-рецессивного типа наследования. Прием фруктозы вызывал у таких больных тошноту, рвоту и одновременно сильное падение содержания глюкозы в крови. Эта форма фруктозурии вызывает задержку физического развития и умственную усталость.
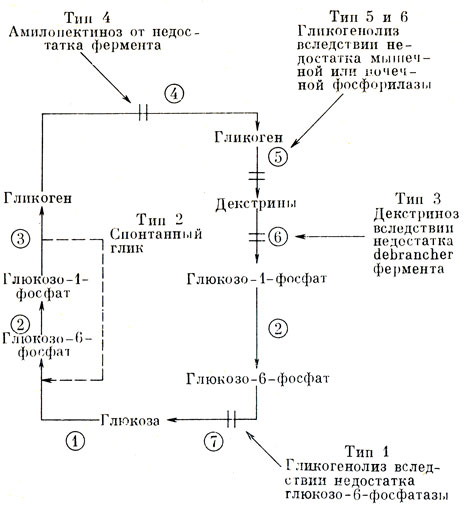
Рис. 38. Цикл обмена гликогена с обозначением метаболических блоков, обусловливающих накопление гликогена 1 - гексокиназа, 2 - фосфоглюкомутаза, 3 - гликогентрансглюкозидаза, 4 - амило-(1,4-1,0) трансглюкозидаза, 5 - фосфорилаза, 6 - амило-1-6 глюкозидаза, 7 - глюкозо-6-фосфатаза
Итак, мы имеем дело с нарушением нормального хода превращений фруктозы. Ну, а каков нормальный ход? Разберемся в нем подробнее, так как фруктоза, которой богаты фрукты, играет немалую роль в нашем питании. Вспомним, что и обычный сахар - сахароза - наполовину состоит из фруктозы.
Полисахариды, содержащие фруктозу, подвергаясь действию ферментов, освобождают ее в кишечнике. Свободная фруктоза переходит из кишечника в кровь, причем по пути приблизительно одна пятая часть ее превращается в глюкозу. Кровь доставляет фруктозу к тканям, клетки которых удивительно быстро расходуют ее. Поэтому содержание фруктозы в крови всегда раз в пятьдесят ниже, чем глюкозы. Совсем недавно были проведены опыты с измерением активности ферментов, принимающих участие в превращении глюкозы и фруктозы. В результате было доказано, что наш организм использует фруктозу лучше, чем глюкозу.
Что же происходит с фруктозой? Вначале то же, что и с глюкозой. Фруктоза - один из поставщиков энергии, столь необходимой для различных проявлений жизнедеятельности организма. Мы знаем, что эта энергия улавливается и конденсируется в макроэргических веществах, обладающих богатыми энергией связями. И тут происходит встреча фруктозы с вездесущей АТФ. В результате такой встречи под воздействием фермента гексокиназы (в присутствии ионов магния) происходит фосфорилирование фруктозы с образованием фруктозо-6-фосфата. Однако эта реакция не всегда осуществима. Дело в том, что гексокиназа тяготеет к глюкозе. Поэтому в присутствии равного количества молекул глюкозы фосфорилирование фруктозы задерживается. В более благоприятных условиях фруктоза как бы находит другой, обходный путь для своих превращений. Здесь уже действует специальный фермент - фосфофруктокиназа, под влиянием которой (с участием АТФ и ионов магния) еще раз происходит фосфорилирование, но уже фруктозо-6-фосфата, который, присоединяя к себе одну частицу фосфорной кислоты, переходит в фруктозо-1, 6-дифосфат.
Теперь позволим себе небольшое отступление. Дело в том, что в основном превращения фруктозы происходят в печени. Конечно, возможно, что и другие органы (в первую очередь мышцы) могут перерабатывать этот сахар. Во всяком случае достоверно известно, что если в распоряжении эритроцитов крови нет глюкозы, то они используют фруктозу. Что касается мышц, то они сначала как бы дают печени превратить фруктозу в глюкозу, а затем уже синтезируют из нее гликоген. Печень так наспециализировалась на переработке фруктозы, что выработала даже особый фермент - фруктокиназу, обеспечивающую моментальное фосфорилирование: присоединение частицы фосфорной кислоты с образованием фрук-тозо-1-дифосфата (при участии АТФ, ионов магния и калия).
В чем же особенность такого фосфорилирования фруктозы? В своеобразной перестраховке. Ведь постоянным и притом всегда побеждающим "соперником" фруктозы на путях обмена веществ является глюкоза, которая как бы мешает ферментам заниматься фруктозой. Но печень имеет в своем распоряжении такую фруктокиназу, которую глюкоза блокировать не может. И если глюкозы много, фосфорилирование фруктозы идет "в обход" именно при участии такой фруктокиназы. (Этот фермент, вероятно, содержится и в мышцах.) Весьма вероятно, что при фруктозурии задерживается реакция, катализируемая именно этой фруктокиназой печени.
Дальнейшие превращения носят довольно сложный характер, и мы на них останавливаться не будем. Отметим только, что они все-таки приводят к образованию глюкозы, пути превращений которой нам уже известны.
Мы уже упоминали, что здоровый организм быстро справляется с фруктозой. Можно проглотить 50 г ее и содержание этого сахара в крови повысится только до 10-15 мг%. Одновременно повышается содержание молочной кислоты в крови, что вполне нормально. Но при фруктозурии такого повышения нет. Зато уровень самой фруктозы в крови может подняться до 70 мг% (после приема этого сахара). Такую форму фруктозурии называют доброкачественной.
Ошибки обмена гликогена. Поскольку речь идет об аномалиях гликогена, уделим внимание этому полисахариду. Имеются основания предполагать, что гликоген не однородное вещество, а смесь нескольких полисахаридов. Достоверно известно, что молекула его сильно разветвлена и построена примерно из 15 цепочек, образованных звеньями - остатками глюкозы, которые связаны между собой кислородными мостиками (рис. 39). В глюкозе, как известно, 6 углеродных атомов. В основной цепи молекулы гликогена кислородные мостики переброшены между первым и четвертым углеродными атомами соседних частиц глюкозы - связь 1 - 4, а боковые цепи образуются, когда эти мостики связывают первый и шестой углеродные атомы - связь 1 - 6.
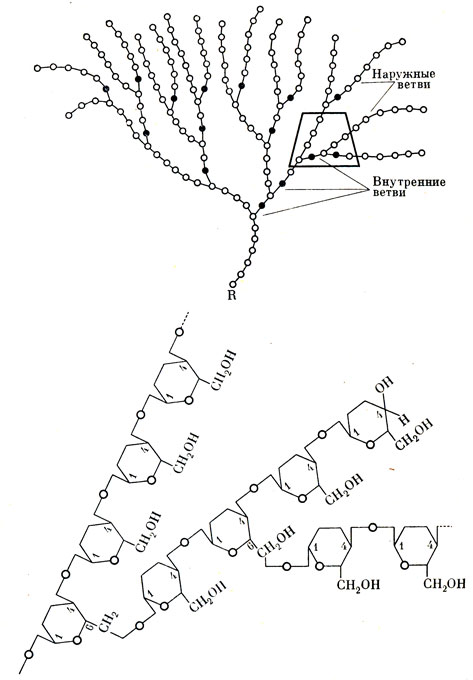
Рис. 39. Модель участка молекулы гликогена. Кружочки обозначают остатки молекул глюкозы, соединенные 1-4 связями, точки - остатки молекул глюкозы, соединенные 1-6 связями. R-кoнечная редуцирующая группа. Внизу - структурная формула участка цепи молекулы гликогена. Связи 1-6 обозначены стрелками, все остальные являются связями 1-4
Теперь понятно, что для синтеза гликогена - наращивания боковых цепочек - важен фермент способствующий превращению связей 1-4 в связи 1-6. А для расщепления гликогена необходимо расчистить путь для фермента фосфорилазы, который наносит удар по связям 1-4. Эту подготовительную работу выполняет особый фермент (амило-1-6-глюкозидаза), который разрывает кислородные мостики в связи 1-6. Теперь путь для фосфорилазы открыт, и она может приступить к расщеплению гликогена, разрывая связи 1-4.
Но содержание гликогена в различных органах и его обмен влияют на многие факторы, из которых один способствуют его накоплению, другие усиливают его распад. К первым относится пища, содержащая много углеводов, особенно фруктозы, действие некоторых гормонов - инсулина, стероидных гормонов надпочечных желез. Ко второй группе можно отнести действие таких гормонов, как адреналин, глюкагон, прогестерон, гормоны гипофиза и щитовидной железы.
В нашем организме гликогеном занимаются несколько ферментов. Их недостаточность (ее можно назвать ферментопатией) может быть обусловлена мутацией соответствующего структурного или контролирующего гена. Полезно познакомиться с этими ферментами поближе. О фосфорилазе мы уже упоминали. Отметим только, что печеночная и мышечная фосфорилазы химически неодинаковы. Запомним также, что недостаточность мышечной фосфорилазы останавливает распад гликогена с самого начала, а это ведет к резкому ослаблению мышц.
Амило-(1-6)-глюкозидаза нам тоже известна. Добавим только, что этот фермент не только высвобождает остатки глюкозы из цепочек гликогена, но и присоединяет к ним воду. При этом образуется свободная глюкоза, которая переходит в кровь.
Фосфоглюкомутаза способствует превращению глюкозо -1-фосфата в глюкозо-6-фосфат.
В печени и почках содержится фермент глюкозо-6-фосфатаза. Его назначение - освобождать глюкозу из глюкозо-6-фосфата. Плохая работа этого фермента может повести к накоплению гликогена в этих органах.
Гексокиназа образует (при содействии АТФ) из свободной глюкозы глюкозо-6-фосфат, а это соединение является ключевым для всех превращений углеводов в нашем теле.
Мы знаем, что цепочка молекулы гликогена разветвлена. Это результат воздействия специального разветвляющего фермента - амило- (1,4 и 2,6)-трансглюкозида-зы, вмешивающегося в процесс синтеза гликогена.
Гликоген в печени и мышцах распадается главным образом при участии фосфорилазы. Этот фермент в тесном сотрудничестве с глюкозо-6-фосфатазой ведает как распадом гликогена с образованием глюкозы, так и синтезом гликогена из глюкозы. Попутно образуется глюкозо-6-фос-форная кислота. Только из этого соединения может освободиться глюкоза, которая переходит затем в кровь. Ясно, что печени необходима глюкозо-6-фосфатаза. А если этого фермента нет или его недостаточно?
Встречается заболевание, при котором отложение гликогена в печени превышает всякие нормы. В результате печень увеличивается, уровень сахара в крови снижается, а рост ребенка задерживается, потому что, хотя глюкоза и усваивается печенью, гликоген теряет способность распадаться или распад его идет крайне медленно из-за недостаточности глюкозо-6-фосфатазы. Эту болезнь описал в 1929 г. Гирке. При болезни Гирке содержание сахара в крови сильно понижено; содержание холестерина и жиров, наоборот, резко повышено. В моче появляется ацетон, что объясняют повышенным разложением жиров в связи с недостатком углеводов. В печени содержание гликогена доходит до 4%, часто повышено содержание жира. Болезнь особенно опасна в первые четыре года жизни. Больных лечат, давая им круглосуточно небольшие количества углеводов, что поддерживает уровень сахара в крови, но не способствует быстрому отложению гликогена. Пища больных должна содержать
Много белков, из которых глюкоза образуется как бы окружным путем. Основной причиной смерти в таких случаях обычно бывают инфекции и ацидоз, возникновение которых необходимо предупреждать. Для лечения инфекций применяют антибиотики и внутривенные вливания глюкозы, ацидоз устраняют с помощью бикарбоната. Хорошие результаты дает применение препаратов тироксина.
Иногда отложение гликогена происходит в мышцах, особенно в сердце. У таких больных большая мышечная слабость. Причиной ее служит недостаток ферментов. Иногда вследствие недостатка фосфорилазы, расщепляющей гликоген, распад этого полисахарида приостанавливается в самом начале. Если недостает фосфорилазы печени, развивается болезнь Херса. Она крайне редка и мало изучена.
Иногда ошибка обмена сказывается в отсутствии трансглюкозидазы, обеспечивающей перенос углеводов при расщеплении молекулы гликогена. Неразветвленная цепочка распадается крайне медленно. В результате гликоген отлагается в печени, селезенке, лимфатических узлах. Болезнь очень трудно распознать. Достоверно известен только один случай, описанный Андерсеном и кончившийся гибелью ребенка. Болезнь Помпе проявляется: в слабоумии, мышечной слабости, расширении сердца, различных нервных расстройствах. Биохимически она прежде всего сопровождается чрезмерным отложением гликогена в сердце, очевидно, вследствие недостаточности фермента, обеспечивающего превращение глюкозы через глюкозо-6-фосфат и глюкозо-1-фосфат в гликоген. Болезнь встречается очень редко (известно менее 30 случаев), но очень тяжела и кончается смертью от недостаточности сердца уже на первом году жизни.
Болезнь Форбса по своим проявлениям напоминает болезнь Гирке, но протекает несколько мягче и встречается еще реже. Ошибка обмена в этом случае выражается в недостаточности амило-1, 6-глюкозидазы, способствующей распаду гликогена, вследствие чего гликоген откладывается в печени, мышцах, сердце. Уровень глюкозы в крови при этом падает. Но поскольку активность глюкозо-6-фосфатазы при этом не затрагивается, организм может использовать глюкозу из других источников. Поэтому больных лечат диетой с высоким содержанием белка, служащего косвенным источником необходимой глюкозы, а жиров дают очень мало. Некоторые ученые рекомендуют также лечение глюкагоном.
Болезнь Мак-Ардля очень редка (известно менее 10 случаев) и характеризуется сравнительно умеренным отложением гликогена в скелетных мышцах, вызванным полной недостаточностью фосфорилазы мышц. Но в печени гликоген не отлагается, так как фосфорилаза функционирует нормально. Пока не выяснено, имеется ли дефект фосфорилазы сердечной мышцы. Болезнь характеризуется болью и резкой слабостью мышц после кратковременной работы. Как известно, в крови здоровых людей после мышечной работы повышается содержание молочной и пировиноградной кислот. В крови больных этой болезнью уровень молочной и пировиноградной кислот после мышечной работы не изменяется. При этой болезни повреждение биохимических систем мешает использованию во время работы запасного "топлива" - глюкозы из мышечного гликогена. При болезни Мак-Ардля больным разрешают только кратковременную физическую работу. По некоторым сведениям, полезно внутривенное введение глюкозы или фруктозы. И, наконец, мышечный гликогеноз отмечается при болезни Сент-Аньезе, очень редкой и малоизученной.
Таким образом, известно семь различных заболеваний, сопровождающихся накоплением гликогена, причем почти в каждом из них обнаружен недостаток особого фермента*. Недавно была открыта болезнь, носящая совершенно "обратный" характер, хотя и здесь замешан гликоген. При этом заболевании почти полностью отсутствует гликоген в печени - агликогеноз. У больных детей (братьев и сестер в одной и той же семье) наблюдается умственная отсталость, резкое падение содержания глюкозы в крови, по утрам судороги, которые можно предотвратить частым и регулярным кормлением ночью. Была проверена активность ферментов, участвующих в обмене гликогена, и удалось напасть на след биохимического повреждения. Ошибка обмена веществ выразилась в отсутствии фермента глюкогенсинтетазы.
*(Число открываемых разновидностей гликогеноза растет. Совсем, недавно описан случай, когда в печени девочки (родители - близкие родственники) совершенно отсутствовал "разветрляющий" фермент.)
Выяснение биохимических нарушений, являющихся причиной молекулярных болезней типа гликогенозов и агликогенозов, дало возможность начать разработку методов молекулярной диагностики и даже молекулярной терапии лечения. Такая диагностика, конечно, затруднена, если отсутствие фермента, ответственного за тот или иной тип заболевания, наблюдается только в печени или мышцах. Но задача облегчается, когда соответствующий фермент отсутствует и в крови, например в ее форменных элементах. А для установления болезни Гирке, т. е. гликогеноза, вызванного отсутствием фермента глюкозо-6-фосфатазы, можно исследовать полость рта. Болезнь Форбса можно распознать по ненормальному строению гликогена в лейкоцитах, болезнь Андерсена - по этому же признаку в эритроцитах. Таким образом, уже имеются более или менее доступные способы химической диагностики. В связи с этим можно отметить, что такие методы разработаны не только для гликогенозов, но и для других наследственных заболеваний.
Пентозурия - выделение с мочой пентоз-моносахаридов с пятью атомами углерода. Можно вызвать пентозурию, обильно питаясь сливами, вишнями или земляникой, которые богаты пентозой. Но так называемая эссенциальная пентозурия представляет хроническое расстройство обмена веществ, не зависящее от содержания пентоз в пище. Это довольно редко встречающаяся врожденная ошибка обмепа аутосомпо-рецессивного типа (один случай на 50 тысяч). Она выражается в ежедневном выделении с мочой 1-4 г пентозы L-ксилулозы. Пентозурия безобидна для организма. Как она возникает, на каких путях обмена совершается ошибка?
Обмен глюкозы может идти разными путями. Тот путь, который ведет к пентозурии, можно назвать побочным, второстепенным. Лица с хронической пентозурией никак от нее не страдают (если не считать того, что их ошибочно считают больными диабетом). Но выделение L-ксилулозы с мочой свидетельствует о том, что где-то произошла ошибка. Где именно? Имеются основания предполагать, что в некоторых случаях начальная реакция превращения глюкозы в глюкозо-б-фосфат на какой-то стадии начинает идти по пути образования глюкуроновой кислоты. Эта кислота и является предшественником L-ксилулозы, так как образует гулоновую, затем кетогулоновую кислоту, из которой уже образуется L-ксилулоза. Здесь цепь превращений обрывается, возможно, из-за наследственного дефекта* в том ферменте, который должен бы превратить L-ксилулозу в ксилитол и т. д. Вместо этого неизменная L-ксилулоза выделяется с мочой.
*(Говоря о наследственном дефекте того или иного фермента, необходимо помнить, что величина активности фермента (как и вообще степень появления любого наследственного дефекта) может сильно различаться. В ряде случаев это может мешать, например, установлению гетерозиготности по мутантному гену. Можно было бы предположить, что такие количественные различия в активности фермента связаны с "дозой" гена. Фактическая проверка не всегда говорит в пользу такого предположения. Однако некоторые исследования показывают, что наследственную недостаточность фермента можно обнаружить в одних тканях (или клетках), а в других тканях она не обнаруживается. Как предполагает Е. Ф. Давиденкова, здесь могут играть роль мутантные изменения не в цельной молекуле фермента, а в одной из субъединиц изоферментов.)
Несколько слов о путях, по которым идут превращения пентоз в нашем организме. Это не столбовая дорога окисления углеводов, а скорее проселочная, которая, казалось бы, не имеет существенного значения. Но к пентозам помимо арабинозы или ксилозы относится также рибоза, и тут же в нашей памяти возникают РНК и ДНК - нуклеиновые кислоты, без которых, как и без белков, нет жизни. Поэтому значение обмена пентоз велико, хотя в количественном отношении он значительно уступает гликолитическому пути окисления углеводов. Именно отсюда, из "пентозного цикла", как его называют, организм черпает рибозу, необходимую для синтеза жизненно важных РНК и ДНК (и для других целей). Здесь же, в пентозном цикле, образуются значительные количества той восстановленной формы пиридиннуклеотида, которая так нужна организму для синтеза жирных кислот.
Разберемся в сущности пентозного цикла. Вспомним, что в процессе гликолиза образуется глюкозо-6-монофосфат. Если к нему присоединится вторая частица фосфорной кислоты, окисление пойдет по обычному гликолитическому пути. А если это не произойдет? Как показал В. А. Энгельгардт, глюкозомонофосфат может не присоединить вторую частичку фосфорной кислоты и пойти по непроторенной дорожке. Здесь он подвергается чрезвычайно сложным превращениям, в дебри которых не будем заводить читателя. Отметим главное: а) глюкозомонофосфат подвергается прямому окислению, отщепляя углеродный атом в виде СО2; б) в ходе превращений образуются пентозы; отсюда и название "пентозный цикл". Но пентоз много. Более того, у совершенно здоровых людей кроме арабинозы и ксилозы в моче могут появиться рибулоза, рибоза и ксилулоза, хотя и в очень небольших количествах. Довольно большие количества рибозы могут выделяться с мочой у лиц с таким заболеванием мышц, которое сопровождается усиленным распадом нуклеотидов (а ведь нуклеотиды содержат в своей молекуле рибозу). Таким образом, ничего "загадочного" в появлении пентозы в моче нет. А вместо термина "пентозурия" было бы правильнее говорить "ксилулозурия", при которой происходит блокада обмена глюкуроновой кислоты. Вина за эту блокаду ложится на рецессивный аутосомный ген.
Хроническая пентозурия не требует лечения. Надо только уметь отличать ее от диабета, выявляя L-ксилулозу в моче при помощи хроматографии. Применение инсулина в этом случае приводит иногда к тяжелым последствиям.
Казалось бы, одни углеводы дают достаточную работу печени. Но этот неутомимый орган трудится и над липидами. Именно печень синтезирует тот холестерин, о котором за последние годы так много пишут на страницах журналов и газет. Его называют то другом, то врагом нашего организма, а то и другом, и врагом одновременно из-за того, что холестерин повинен в возникновении атеросклероза.
Мы уже упоминали о том, что холестерин - необходимая и очень важная составная часть клеток тела человека, особенно мембран. Кроме того, холестерин занимает важное положение в качестве исходного строительного материала для синтеза других стероидов. Именно в печени, не довольствуясь холестерином пищи, наш организм строит свой собственный холестерин, во всяком случае большую его часть, которая путешествует с плазмой крови и может откладываться в стенках артерий. Печень же связывает холестерин с жирными кислотами, в результате чего образуются эфиры холестерина. Печень строит и своеобразные транспортные средства - фосфатиды (главным образом лецитин), с помощью которых холестерин путешествует в плазме крови.
В печени происходит окисление холестерина и превращение его в желчные кислоты, часть которых связывается аминокислотой - глицином и таурином. Печень выделяет желчные кислоты с желчью, после чего большая часть их всасывается обратно. В обмене липидов печень играет главную роль. В отношении простых липидов (жиров) у нее имеется соперник - жировая ткань. Но это друзья-соперники: между ними происходит активный обмен липидов. Жировая ткань, как и печень, способна синтезировать жирные кислоты, а так как жировой тканью наше тело богато (в ней в пять раз больше жира, чем во всей остальной массе тела), то, естественно, с ее ролью в обмене липидов нельзя не считаться. Печень синтезирует свои липиды и при нормальных условиях откладывает часть их в жировой ткани. А в результате голодания при появлении ацетоновых тел (последствие обеднения печени гликогеном и сахарного диабета) и различных повреждений преобладает обратный процесс, ведущий к ожирению печени*. Это заболевание печени заслуживает внимания.
*(Ученые предполагают, что поджелудочная железа вырабатывает особый гормон - липокаин, предохраняющий печень от ожирения.)
К ожирению печени предрасполагают и даже вызывают его множество факторов: некоторые гормоны (гормон роста), яды, инфекционные болезни, качество питания (недостаток ненасыщенных жирных кислот и веществ, богатых метильными группами - холина, метионина). Организм человека располагает ограниченным количеством метильных групп и периодически остро нуждается в этих доставляемых пищей "частичных витаминах". Пища содержит иногда избыток таких веществ, которые могут отнимать дефицитные метильные группы, что опять-таки способствует ожирению печени. Наконец, кислородное голодание тоже ведет к ожирению печени.
Поскольку обмен витамина А тесно связан с жирами, к печень является главным складом, хранящим большой запас этого витамина и его провитамина - каротина.
Печень - главный распорядитель в белковом хозяйстве человеческого организма. Прежде всего только в печени образуется мочевина, а ведь это вещество - продукт, с которым выделяется белковый азот из нашего организма. Несмотря на малую величину молекулы мочевины, образование ее в нашем теле требует значительной затраты энергии, в чем, конечно, принимает участие опять та же АТФ. Как и многие реакции в организме, процесс этот носит циклический характер. Начало свое мочевина берет от аминокислот, поскольку в ней содержатся две аминогруппы, поставщиками которых могут служить любые аминокислоты. Одна из них - глютаминовая кислота - доставляет аминогруппу к специальной аминокислоте - цитруллину (он образуется с помощью АТФ из другой аминокислоты - орнитина). Так начинается цикл ферментативных превращений. Цитруллин образует аминокислоту - аргинин,- содержащую остаток мочевины. Фермент аргиназа высвобождает мочевину. Причем снова образуется орнитин, и продолжается цикл химических превращений, ведущих опять-таки к образованию мочевины, которая затем переносится кровью к почкам и выделяется с мочой.
Печень расноряж:ается судьбой многочисленных аминокислот, из которых состоят белки. Такое разнообразие аминокислот не всегда соответствует потребности организма, поэтому в ней выработалась способность переводить одну аминокислоту в другую. Это осуществляется путем переноса специфической для аминокислот аминогруппы на какую-либо кислоту. В результате образуется определенная аминокислота.
Реакцию переноса аминогруппы называют трансаминированием (переаминированием), а катализирующие эту реакцию ферменты - трансаминазами. Так, например, войдя в эту реакцию, глютаминовая аминокислота и пировиноградная кетокислота выходят из нее, превращенные в аминокислоту аланина и кетоглютаровую кетокислоту.
Печень может переделывать аминокислоты и другим способом. Действуя специальным ферментом (главным образом глютаматдегидрогеназой), она превращает аминокислоты в кетокислоты, освобождая аминогруппу в виде аммиака, который тут же используется для синтеза мочевины. Да иначе и быть не может: аммиак - враг тканей нашего тела, и его необходимо обезвреживать.
Печень влияет на метаморфоз других азотистых веществ - на синтез креатина и холина - соединений, играющих важную роль в обмене веществ. В печени, наконец, происходит синтез важнейших белков плазмы крови, в том числе и таких, которые участвуют в ее свертывании, - протромбина и фибриногена. В этом случае печень особенно уязвима и даже при незначительных воздействиях может делать ошибки.
В печени осуществляется и один из конечных этапов распада нуклеиновых кислот, ведущий к образованию мочевой кислоты, которая затем выделяется с мочой.
По "совместительству" печень занимается и такими жизненно важными для нашего тела пигментами, как гемоглобин: в ней в основном осуществляется распад. Она постепенно расщепляет гемоглобин с помощью соответствующих ферментов и под конец отщепляет белок и железо, накапливая его в виде ферритина. Остается красящее вещество билирубин, которое в печени соединяется главным образом с глюкуроновой кислотой.
Почти 90% всего билирубина образуется из гемоглобина, остальные 10% из других источников. Билирубин нерастворим, и это не позволяет ему самостоятельно выходить из клеток ретикуло-эпдотелиальной системы (система особых клеток, рассеянных в разных органах и имеющих защитное значение). Следовательно, он нуждается в транспортных средствах. Эти средства ему предоставляет плазма крови в виде особых белков - альбуминов и глобулинов. Они присоединяют билирубин и, таким образом, выводят из клеток. В крови его содержится немного, не больше 10 мг в литре плазмы крови.
Но и в крови билирубин не растворяется. Только соединение с другими веществами позволяет ему переходить в растворимую форму. Главное из этих веществ - глюкуроновая кислота. Обычно билирубин соединяется с двумя молекулами глюкуроновой кислоты (иногда достаточно и одной), образуя диглюкуронид билирубина. И тут не обходится без вмешательства ферментов. Сначала идет подготовка глюкуроновой кислоты. Исходным сырьем служит уридиндифосфатглюкоза. Это особое соединение атакует фермент дегидрогеназа, переводя его в уридиндифос-фатглюкуроновую кислоту. Итак, глюкуроновая кислота имеется, но ее нужно соединить с билирубином. Эту задачу выполняет другой фермент - трансфераза. Билирубин, соединенный с глюкуроновой кислотой, переходит затем в желчь и в пищеварительный канал, где подвергается дальнейшим превращениям в уробилиноген и стеркобилиноген.
Путь из крови в желчные протоки тернист. Его контролируют различные гены, вырабатывающие соответствующие ферменты. Ферменты нужны для проникновения билирубина в печеночные клетки, для размещения его в митохондриях этих клеток и для глюкуропирования и выхода билирубина в желчные капилляры. И так как гены могут совершать ошибки, недостаточно синтезируется тот или ипой фермент или образуются ферменты-уроды. В результате билирубин накапливается в крови (гипербилирубинемия) и возникает желтуха. Некоторые формы желтухи могут быть наследственными. Приведем несколько примеров таких наследственно обусловленных расстройств работы печени.
Как различать билирубины, связанные и не связанные с глюкуроновой кислотой? Очень просто. Каждому клиницисту хорошо знакома реакция Ван-ден-Берга с диазореактивом. Билирубин, соединенный с глюкуроновой кислотой (связанный билирубин), растворим в воде и поэтому дает прямую реакцию с диазореактивом. Но билирубин, не соединенный с глюкуроновой кислотой (свободный билирубин), дает с диазореактивом непрямую реакцию только после добавления алкоголя.
Гипербилирубинемия новорожденных обычно приводит к гибели уже в детстве. Она наследуется по рецессивному типу. Эта гипербилирубинемия не связана с глюкуронидами. Ошибка заключается в недостатке специфических ферментов - дегидрогеназы и трансферазы.
Врожденная негемолитическая желтуха тоже тяжелое заболевание. Если дети и выживают, то страдают мозговыми расстройствами и общим недоразвитием. Ошибка обмена здесь не только в ферментах, связывающих билирубин с глюкуронидами, но и в неполноценности самой молекулы билирубина.
Болезнь Жильбера также наследственное расстройство функции печени, передающееся по доминантному типу. У мужчин оно встречается в 10 раз чаще, чем у женщин. Протекает хронически и обычно легко. Реакция Ван-ден-Берга непрямая. Ошибка заключается в неспособности клеток печени обеспечить соединение билирубина с глюкуронидами. Болезнь, будучи генетической, развивается только после появления вторичных половых признаков в юношеском возрасте. Очевидно, имеется какая-то не выясненная пока связь мутировавшего гена с половой гормональной активностью организма.
Гипербилирубинемия с повышением содержания прямого билирубина наследуется доминантно, аутосомно Протекает легко. Ошибка в нарушении транспорта билирубина, связанного в печеночных клетках.
Наконец, изредка встречаются и другие формы наследственной гипербилирубинемии, связанной с глюкуронидами и с прямой реакцией Ван-ден-Берга.
Таким образом, существует несколько видов наследственной билирубинопатии, различающихся и по типу наследования, и по характеру нарушений билирубиновогс обмена.
Образование желчи - очень важная функция печени. Ведь желчь не только помогает пищеварению и всасыванию липидов, с ней выделяются конечные продукты обмена и выводятся чуждые организму вещества. Наконец, в печени осуществляется взаимосвязь между обменом углеводов, жиров, белков.
Печень играет важную роль, обезвреживая чужеродные вещества и соединения нашего организма. Но, к сожалению, она может и ошибаться. Бывают случаи, когда безвредные вещества, попадая в печень, становятся ядовитыми. Но в общем печень направляет свои ферменты на защиту организма от вредных веществ. Она действует совместно с кишечником. Большая часть веществ, которые образуются в кишечнике под действием микробов, попадает в печень, а оттуда после обезвреживания снова выделяется в кишечник.
Липиды - общее название для жиров и жироподобных веществ - липоидов. Нейтральным жиром химики называют сложный эфир, в котором одна молекула глицерина соединена с тремя молекулами жирных кислот. Жироподобные вещества тоже сложные эфиры с той, однако, разницей, что в некоторых из них глицерин соединяется не только с жирными кислотами, но и с другими веществами, в частности с фосфорной кислотой. Такие липоиды носят название фосфатидов. В других липоидах - стеридах - жирные кислоты соединены но с глицерином, а с другими, более сложными спиртами вроде холестерина.
Остановимся более подробно на судьбе липидов в межуточном обмене. Она несколько отличается от превращении белков и углеводов, которые кишечные ферменты дробят па мелкие осколки, переходящие в кровь. Во-первых, жиры перевариваются по полностью, и часть их в виде очень мелких капелек непосредственно всасывается степкой кишок. Во-вторых, большая часть жиров разлагается кишечными ферментами на глицерин и жирные кислоты, но уже в клетках кишечного эпителия из них снова синтезируются жиры. В-третьих, те и другие жиры переходят через кишечную стенку (здесь фосфорная кислота частично превращает их в фосфолипиды) сначала в лимфатические сосуды, и только потом через грудной лимфатический поток они поступают в кровоток.
Кровь сначала доставляет жиры в подкожную жировую клетчатку и сальники и откладывает их здесь про запас. В эти склады поступают и те жиры, которые клетки организма синтезируют из продуктов распада углеводов и некоторых аминокислот. Уже отсюда кровь доставляет жиры к местам их использования, главным образом в клетки печени.
Специальные ферменты клетки - липазы - разлагают жиры на глицерин и жирные кислоты. Дальнейшая судьба глицерина похожа на судьбу глюкозы. Его превращения начинаются с вмешательства АТФ, а кончаются распадом до молочной кислоты с последующим окислением до углекислого газа и воды. Реже происходит синтез из него гликогена - главного представителя углевода в нашем теле. Здесь мы видим еще одну связь между обменом жиров и углеводов.
Но превращение жирных кислот клетка ведет по совершенно другому пути. Под действием множества ферментов и, конечно, при вмешательстве АТФ она "отщипывает" от жирных кислот по два атома углерода в виде уже знакомой нам активной уксусной кислоты, которую затем превращает в ацетоуксусную кислоту. Это происходит главным образом в клетках печени. Ацетоуксусную кислоту захватывает затем кровь и доставляет к клеткам различных тканей и органов, которые и разлагают ее на конечные продукты - углекислый газ и воду. Такова судьба насыщенных жирных кислот. Что касается ненасыщенных (которыми богаты растительные жиры), то их превращения в клетках организма пока еще изучены недостаточно. Мы знаем уже, что некоторые из них - линолевую, линоленовую и арахидоновую - клетки нашего тела синтезировать не могут. А они очень нужны для нормального обмена жиров.
Обмен фосфатидов в нашем организме тесно связан с обменом жиров. Фосфатиды помогают всасыванию жиров, участвуют в их переносе кровью и предотвращают ожирение печени. Они играют важную роль в органах размножения и при развитии зародыша. Клетки нашего тела (особенно печени) постоянно обновляют состав фосфатидов, меняя входящие в их состав жирные кислоты. Клетки располагают целой армией специальных ферментов, разлагающих фосфатиды, конечно, с участием АТФ. Судьба отщепляющихся при этом глицерина и жирных кислот нам уже известна. А вот главная азотистая составная часть фосфатидов - холин - настолько необходима клеткам, что многие ученые считают его витамином. Достаточно сказать, что часть холина идет на образование ацетилхолина - вещества, необходимого для передачи нервных импульсов.
Наследственные болезни обмена липидов затрагивают тзличные стороны превращений этих веществ. Их можно разделить на две группы: 1) первичные семейные липидозы, для которых характерно повышение содержания липидов (гиперлипемия) в крови, и 2) семейные липидозы, характеризуемые накоплением сложных липидов - сфинголипидов - внутри клеток.
Остановимся на болезнях первой группы. Более 30 лет назад немецкие врачи Бюргер и Грюти обратили внимание га больного с увеличенной печенью и селезенкой. Для болезни были характерны приступы острых болей в животе, особенно после принятия жирной пищи, и появление на коже ксантом - желтых опухолей, повышенная мутность плазмы крови. В крови оказалось сильно повышено одержание жиров - гиперлипемия - в виде мелких капелек - хиломикронов. Болезнь, в общем, протекала легко. Наследственный ее характер и преимущественно доминантного типа был установлен несколько позднее и прослежен приблизительно в 40 случаях, описанных в литературе. В некоторых случаях удалось установить наследственный дефект обмена в липазе липопротеинов. В других случаях предполагаются иные нарушения обмена, в частности блокада обмена жиров и замедленное выведение хиломикронов плазмы крови.
Гиперлипемия наследуется так, что у гомозиготов проявляется более тяжело, а у гетерозиготов только в виде тенденции к гиперлипемии и атеросклерозу. Таким образом, соответствующий ген в гетерозиготном состоянии определяет лишь предрасположенность к гиперлипемии, а в гомозиготном состоянии - к тяжелому наследственному заболеванию.
Советскими учеными твердо установлено, что атеросклероз и гипертоническая болезнь встречаются гораздо чаще среди кровных родственников, чем в семьях здоровых лиц. Необходимо учитывать, что повышение содержания холестерина или липидов в крови - это только одно из проявлений атеросклероза и гипертонии. Если же взять эти заболевания в целом, то наследственное предрасположение к ним, вероятно, обусловливается многими генами. Одни из них ведущие, другие второстепенные, но каждый из них выводит из строя различные ферментные системы, например, участвующие в свертывании крови.
По современным взглядам, в крови человека есть две системы: система свертывающих веществ и их антагонистов - противосвертывающих веществ. Взаимоотношения между этими двумя системами регулируются нервной системой, которая следит за равновесием всех свертывающих веществ крови. Преобладание в крови любого из этих веществ одинаково неблагоприятно для организма. Поэтому, как только намечается нарушение равновесия, следует приказ нервной системы о выработке дополнительных количеств веществ противоположного действия для поддержания баланса свертывающих и противосвертывающих факторов крови. Механизм этой регуляции зависит еще и от множества других явлений и причин, изучение которых продолжается в настоящее время, И здесь ученые снова встретились с атеросклерозом. Оказалось, что противосвертывающие вещества - антикоагулянты - вырабатываются в организме стенками сосудов. А при атеросклерозе уменьшается выработка стенкой сосуда веществ, предупреждающих свертывание. Именно этим, как указывают современные исследования, и объясняется столь частое, но вовсе не обязательное сочетание атеросклероза с инфарктом миокарда. Борьба с ожирением и сведение жира в пище до минимума (30-50 г в сутки) позволяют с успехом лечить таких больных.
Другая болезнь этой группы характеризуется высоким содержанием холестерина и фосфолипидов в крови. Ее называют эссенциальной семейной гиперхолестеринемией. При этом часто развиваются ксантомы кожи и сухожилий и, что очень серьезно, обнаруживается склонность к быстрому развитию атеросклероза. Болезнь наследуется по аутосомно-доминантному типу. Ошибка обмена веществ заключается в механизме, регулирующем концентрацию холестерина в крови, но точная природа ее пока не выяснена. Совсем недавно установлена новая форма этого заболевания, причиной которого является недостаточность в плазме крови особого фермента - лецитин-холестеринацилтрансферазы.
Предложены различные виды лекарственного лечения: гормональные препараты, никотиновая кислота и др., но все они сопряжены с нежелательными последствиями. Больным можно только рекомендовать умеренно питаться, избегать продуктов, богатых насыщенными жирными кислотами (сало, мясо, яйца), и заменять их растительными маслами с высоким содержанием ненасыщенных жирных кислот (кукурузное, арахисовое масло), хотя это не всегда достигает цели.
Что касается наследственных липидозов второй группы, то их несколько.
Амавротическая (по-латыни "амаурос" - темный, слепой) идиотия появляется обычно на первом году жизни. Характеризуется вялостью, апатией, нарастающим слабоумием, падением зрения до слепоты, мышечной слабостью, параличами. Кончается гибелью к концу второго - началу третьего года. Характерный признак - вишнево-красное пятно в глазном дне. Это наследственная болезнь аутосомно-рецессивного типа. Известно более 300 случаев, причем в большинстве случаев обнаружено кровное родство между родителями.
Биохимическое повреждение состоит в дефекте фермента (какого именно, пока не установлено), участвующего в обмене ганглиозидов в клетках ганглиев. Ганглиозиды настолько заполняют клетки, что вызывают их гибель. Характерная составная часть ганглиозидов - сиаловая, или нейраминовая, кислота, наряду с которой входят гексозы, жирная кислота, хондрозамин и сфингозин.
Известны "позднедетская" и юношеская формы этой болезни. Юношеская форма проявляется в 5-8 лет прогрессирующим ослаблением зрения и умственных способностей. Долголетняя болезнь ведет к слепоте, идиотизму и кончается смертью. Эта форма особенно распространена в Швеции, где частота ее определяется 1:400. В отличие от детской формы эта болезнь может быть следствием другой мутации. Совсем недавно были сделаны попытки лечения введением в кровь особой смолы, понижающей содержание нейраминовой кислоты и холестерина. Но это не улучшило состояние больных.
В болезни Нимана - Пика липидоз обусловлен главным образом накоплением в ретикуло-эндотелиальной и других тканях сфингомиелина, содержание которого в печени и селезенке в десятки раз превышает нормальное. Очевидно, ошибка обмена затрагивает превращение сфингомиелина, но на каком именно участке, пока не выявлено. Здесь возможны: а) избыточный синтез сфингомиелина; б) образование "изуродованных" молекул сфингомиелина, на которые ферменты не могут действовать; в) недостаток ферментов, расщепляющих сфингомиелин. Совсем недавно найдено, что при этой болезни сильно понижено содержание сфингомиелина в эритроцитах крови, тогда как при амавротической идиотии этого нет. Эта болезнь чаще встречается у детей, родители которых состоят в кровном родстве. Она носит рецессивный характер. Известно около 100 случаев, главным образом в еврейских семьях. Для лечения испробованы переливание крови, удаление селезенки, массивные дозы витамина А, кортизон, липотропные вещества и другое, но без заметного эффекта. Болезнь начинается на первом году жизни и кончается гибелью ребенка к трем годам.
К этой же группе относится болезнь Гоше, при которой находят отложения цереброзидов* в характерных крупных клетках Гоше. Она протекает тяжело в детском возрасте, когда сопровождается разрушениями нервной системы, легче - у взрослых. В большинстве случаев наследуется по аутосомно-рецессивному типу, в литературе описано около 150 случаев, аномалия обмена находится в механизме, регулирующем отложение цереброзидов в клетках. Однако точных сведений пока что нет. Биохимически болезнь всегда сопровождается повышением активности сывороточной кислой фосфатазы, но причины этого не выяснены. Из методов лечения наибольший эффект дает удаление селезенки, в которой накапливаются цереброзиды. Боли успокаиваются после приема стероидных препаратов.
*(Цереброзиды - это липиды, содержащие сфингозин (аминоспирт), различные жирные кислоты и галактозу (или глюкозу).)
Болезнь Фабри - редкое, генетически обусловленное необычайно большое накопление некоторых гликолипидов в тканях. Хотя биохимически она изучена, но место дефекта пока не обнаружено.
Мало изучены и липоидозы, при которых в печени и других органах скапливаются сульфогликолипиды. Известно менее 300 случаев другой наследственной болезни этой группы - гаргоилизма (от французского слова "гаргуй" - рыльце водосточной трубы готических соборов, украшенных фигурами с уродливыми лицами). При этой болезни, главным образом аутосомно-рецессивной формы, явно нарушен обмен ганглиозидов и свободных мукополисахаридов, но место ошибки обмена пока не выяснено.
Совсем недавно ученые предположили, что вина ложится на гены, контролирующие ферментную систему лизосом, не справляющуюся при гаргоилизме с построением нормальной молекулы гепаринсульфата. Из мочи больных выделили гепаринсульфат, молекула которого резко отличалась от гепаринсульфата из нормальной мочи. Впрочем, другие ученые, изучая легкие случаи гаргоилизма без умственной отсталости, считают, что умственное недоразвитие мало связано с выделением гепаринсульфата. Содержание этих веществ заметно повышено в мозге, селезенке, печени, лимфатических узлах. Внешний вид больных очень характерен. Лечепие симптоматическое, но гаргоилизм пока неизлечим. Комбинированное лечение облучением гипофиза и тиреоидином дает только временное улучшение.
Недавно описаны случаи, которые хотя и относят к этой же группе заболеваний, но можно с успехом считать расстройствами углеводного обмена. Это тяжелое наследственное расстройство, первопричина которого лежит в гене, ведающем ферментом бета-галактозидазой. Фермент этот не справляется с основной своей задачей - отщеплением галактозы из мукополисахаридов.
Расстройство обмена липидов происходит и при болезни Рефсума. В начале этой главы мы напоминали, что окисление жирных кислот с неразветвленной цепью происходит по типу бета-окисления, т. е. распадается не у первого альфа-углерода, но у второго - бета-углерода. Если же цепь жирной кислоты разветвлена, то в этом случае у нормальных людей процесс распада начинается у альфа-углерода, а потом уже идет бета-окисление. При болезни Рефсума этот механизм нарушен, в результате чего в крови и жировой ткани происходит образование совершенно необычной для нашего тела фитановой кислоты с очень длинным химическим названием - 3, 7, 11, 15-тетраметил-гексадекановая кислота. Распадаясь, эта кислота образует углеводороды - фитан и триэтан, которые содержатся в жировой ткани.
При использовании материалов сайта активная ссылка обязательна:
http://anfiz.ru/ 'Анатомия и физиология человека'